
One of the main aims of our research is to elucidate the molecular basis of neurodegeneration and were therefore inspired to embark on a screen utilizing fly photoreceptors to isolate genes required for neuronal maintenance. This extensive screening effort isolated 700 mutations corresponding to 165 genes (Yamamoto et al., 2014). The results of this screen have provided a rich resource of novel mutants for the fly community and have permitted us to dissect mechanisms for a variety of diseases.
Alzheimer's Disease (AD)
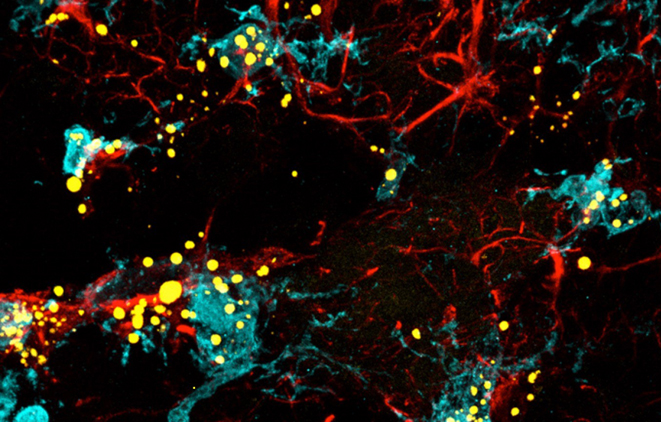
By performing TEM on eye clones of numerous mitochondrial mutants isolated in this screen, we discovered a dramatic increase in Lipid Droplets (LD) in glial cells (pigment cells) of some mutants. Several of these mutants exhibit elevated ROS with accumulated peroxidated lipids in glia. This elevated ROS associated with mitochondrial dysfunction triggers c-Jun-N-terminal Kinase (JNK) and Sterol Regulatory Element Binding Protein (SREBP) activity in neurons, leading to the synthesis of lipids. These lipids are then shuttled to glia to form LD that are rich in peroxidated lipids, which lowers ROS in neurons and delays the onset of neurodegeneration. Our model suggests that delay of neuronal demise is likely the result of ROS sequestration in LD. This phenomenon is evolutionarily conserved as it is also observed in mice that lack Ndufs4 (a subunit of Complex 1). Just as in the fly model, these mice accumulate LD in astrocytes and microglia prior to neural loss and death (Liu et al., 2015).
The neuronally produced lipids are transferred to extracellular apolipoproteins like ApoD and ApoE, which are critical for glial LD formation (Liu et al., 2017). Human ApoE3 can substitute for the fly glial apolipoprotein ApoD. However, ApoE4, the most prominent Alzheimer's Disease (AD) susceptibility allele, impairs lipid transport and LD formation because it likely cannot be properly lipidated. In contrast, ApoE2, a neuroprotective allele, leads to an efficient accumulation of peroxidated LD in glial cells and protects against neurodegeneration.
We then studied numerous genes identified in Genome Wide Association Studies for AD in an attempt to pinpoint where some of the genes/proteins fit into the pathway (Moulton et al., 2022). We tested the role of some AD risk genes in ROS-induced LD formation and showed that several genes/proteins impact neuroprotective LD formation, including homologs of human ABCA1, ABCA7, VLDLR, VPS26, VPS35, AP2A, PICALM, and CD2AP. We also showed that ROS enhances Aβ42 phenotypes in flies and mice. Interestingly, a peptide agonist of ABCA1 restores glial LD formation in a humanized APOE4 fly model, a potentially therapeutic avenue.
Tau is another major player in AD. We recently found that loss or overexpressing Tau in glia disrupts LDs in flies and rat neuron–astrocyte co-cultures, sensitizing glia to ROS (Goodman et al., 2024). We discovered that endogenous Tau is required for glial LD formation from the ER and that normal Tau protects against neuronal ROS that induces peroxidated lipid production in neurons. The peroxidated lipids accumulate in the glial ER and cannot properly form LD when Tau is aberrant as the formation of the LD require microtubules whose function is regulated by Tau. Flies that lack glial Tau have decreased lifespans and motor defects that are rescuable by administering the antioxidant N-acetylcysteine amide. Overall, this work provides insights into the important role that Tau has in glia to mitigate ROS in the brain.
Parkinson's Disease (PD)
We embarked on the study of iPLA2-VIA, a gene whose loss causes infantile neuroaxonal dystrophy (early death) as well as early-onset parkinsonism. Loss of the fly homolog, PLA2G6, reduces lifespan to less than 30 days (normally ~90 days), impairs synaptic transmission, and causes the demise of the CNS and the visual system. Phospholipases typically hydrolyze glycerol phospholipids, but loss of iPLA2-VIA does not affect phospholipid composition in the brain but rather causes an elevation in ceramides. We discovered that PLA2G6 binds two retromer subunits, Vps35 and Vps26, and is required to maintain proper retromer function. Loss of PLA2G6 causes a progressive loss of retromer function and leads to a deficit in the recycling of proteins and membranes from endosomes to plasma membrane, thereby increasing trafficking from the endosome to the lysosome. This leads to progressive lysosomal expansion and ceramide accumulation caused by lysosomal dysfunction. As ceramide increases and stiffens the membrane a feedback loop then impairs endocytosis and retromer function, further expanding lysosomes and elevating ceramide. A severe reduction in the levels of Vps35 and Vps26, enlargement of lysosomes, and an increase in ceramides is also observed upon overexpression of alpha-synuclein (Lin et al., 2018; 2022). These defects may be shared in Parkinson disease (PD) as synuclein accumulation is a common hallmark in PD patients. We are currently exploring how this pathway interacts with other genes associated with PD, including GBA. We have shown that glucosylceramide is produced in neurons and degraded by Gba1b in glia (Wang et al., 2022). Loss of Gba1b leads to lysosomal expansion and glucosylceramide accumulation, resembling the phenotypes we observed in iPLA2-VIA mutants. We argue that the lysosomal ceramide metabolism pathway is important for the pathology of PD.
Lin G, Lee PT, Chen K, Mao D, Tan KL, Zuo Z, Lin W-W, Wang L, Bellen HJ (2018) Phospholipase PLA2G6, a parkinsonism-associated gene, affects Vps26 and Vps35, retromer function, and ceramide levels, similar to a-Synuclein gain. Cell Metabolism 28:605-618. PMID: 29909971.
Lin G, Tepe B, McGrane G, Tipon RC, Croft G, Panwala L, Hope A, Liang AJH, Zuo Z, Wang L, Bellen HJ (2023) Exploring therapeutic strategies for Infantile Neuronal Axonal Dystrophy (INAD/PARK14).eLife :doi:10.7554/eLife.82555. PMID: 36645408.
Liu L, MacKenzie KR, Putluri N, Maletic-Savatic M, Bellen HJ (2017) The glia-neuron lactate shuttle and elevated ROS promote lipid synthesis in neurons and lipid droplet accumulation in glia via APOE/D. Cell Metabolism 26:719-737. PMID: 28965825 . Covered in Cell Metabolism 26(5):701-2, Preview, and Science Translational Medicine 9(412), eaap8170, Editor's Choice.
Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, Bellen HJ (2015) Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 160:177-190. PMID: 25594180.
Moulton MJ, Barish S, Ralhan I, Chang J, Goodman LD, Harland J, Marcogliese PC, Johansson JO, Ioannou M, Bellen HJ (2021) Neuronal ROS-Induced Glial Lipid Droplet Formation is Altered by Loss of Alzheimer's Disease-associated Genes. Proceedings of the National Academy of Sciences 118(52):e2112095118. PMID: 34949639.
Wang L, Lin G, Zuo Z, Li Y, Byeon SK, Pandey A, Bellen HJ (2022) Neuronal activity induces Glucosylceramide that is secreted via exosomes for lysosomal degradation in glia. Science Advances 8(28):eabn3326. PMID: 35857503.
Yamamoto S, Jaiswal M, Charng WL, Gambin T, Karaca E, Mirzaa G, Wiszniewski W, Sandoval H,Haelterman NA, Xiong B, Zhang K, Bayat V, David G, Li T, Chen K, Gala U, Harel T, Pehlivan D, Penney S, Vissers L, de Ligt J, Jhangiani SN, Xie Y, Tsang SH, Parman Y, Sivaci M, Battaloglu E, Muzny D, Wan YW, Liu Z, Lin-Moore AT, Clark RD, Curry CJ, Link N, Schulze KL, Boerwinkle E, Dobyns WB, Allikmets R, Gibbs RA, Chen R, Lupski JR, Wangler MF, Bellen HJ (2014) A Drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 159:200-214. PMID: 25259927. Recommended by F1000. Covered in Nature Methods 11(12):1197, Comment.